7.7. BaTiO3 : An SCPH-based structural optimization example
This page explains how to calculate crystal structures at finite temperatures based on the SCPH theory. We calculate the cubic-tetragonal structural phase transition of BaTiO3. We fix the shape of the unit cell and calculate the temperature(\(T\))-dependence of the atomic positions.
The example input files are provided in example/BaTiO3/scph_relax.
Let’s move to the example directory
$ cd ${ALAMODE_ROOT}/example/BaTiO3/scph_relax
1. Prepare force constants
This tutorial assumes that the harmonic and anharmonic force constants are already calculated up to the fourth order. Please copy the file of IFCs calculated in Tutorial 7.5 to the current directory.
$ cp ../anharm_IFCs/4_optimize/reference/cBTO222.xml.zip ./
$ unzip cBTO222.xml.zip
Note
We need to calculate the force constants in the phase with the highest symmetry (cubic phase in the case of BaTiO3) to calculate the structural phase transitions. This is because the calculated set of IFCs satisfies the symmetry at this reference structure.
Suppose we calculate the IFCs at orthorhombic structure, for example. In that case, the generated IFCs do not satisfy the symmetry between the states with opposite polarizations, and the structure will not converge to the high-symmetry cubic phase at high temperatures.
2. Prepare the input file
In addition to the input file of the SCPH calculation at the fixed reference structure
(See Tutorial 7.4 for example),
we need to set RELAX_STR-tag in &scph-field, &relax-field, and &displace-field properly.
We specify the initial atomic displacements in &displace-field,
which are added to the high-symmetry reference structure.
This is necessary to induce spontaneous symmetry breaking from the cubic phase.
The input file of the anphon calculation is BTO_scph_thermo.in. The lines
SET_INIT_STR = 3
COOLING_U0_INDEX = 5
COOLING_U0_THR = 0.005
are for the cooling calculation.
With SET_INIT_STR = 3, the initial structure of the SCPH-based structural optimization
is set from &displace-field if the structure at the previous temperature converges to the
high-symmetry phase.
The structure is considered to be in the high-symmetry phase if the COOLING_U0_INDEX th component
of the atomic displacement is smaller than COOLING_U0_THR [Bohr].
Because we count the components from zero, COOLING_U0_INDEX = 5 means that we focus on
the \(z\)-component of the second atom (Ti).
Please see the documentation for more detailed explanations.
To perform the heating calculation, set LOWER_TEMP = 0 in &scph-field and SET_INIT_STR = 2.
Then, write the low-temperature structure to the &displace-field.
Note
We use a coarse SCPH \(q\)-mesh of KMESH_SCPH = 4 4 4 to save computational cost.
Convergence with respect to KMESH_SCPH and the threshold COORD_CONV_TOL needs
to be carefully checked to obtain accurate calculation results.
Note
The convergence of the structure gets significantly slower right at the vicinity of the phase transition because the gradient of the free energy almost vanishes. In such cases, getting a smooth \(T\)-dependence for materials with more complicated structures is sometimes difficult.. This problem can be partially avoided by choosing a larger \(T\)-step and estimating the transition temperature from the crossing point of the free energies with different phases.
Now, run the calculation with
$ ${ALAMODE_ROOT}/anphon/anphon BTO_scph_thermo.in > BTO_scph_thermo.log
Note
The calculation can takes more than 10 minutes if you don’t use parallelizations.
If you want to try the calculation in a shorter time, please use a larger value for
COORD_CONV_TOL = 1.0e-5 or DT = 25,
or make TMAX = 400 smaller.
The structure will not be completely convergent for large COORD_CONV_TOL,
but you will be able to get the overview of the calculation.
The calculation time may be shorter in the future as we will implement a more sophisticated algorithm for the structure update.
3. Analyze the calculation results
Plotting the result with
$ gnuplot plot_structure.plt
you will get the following plot.
The atomic displacements are zero at high temperatures, where the structure converges to the high-symmetry cubic phase. At low temperatures, the atoms are displaced along the \(z\)-direction, and the structure is in the tetragonal phase. The estimated transition temperature (\(T_c\)) is around 150~175 K.
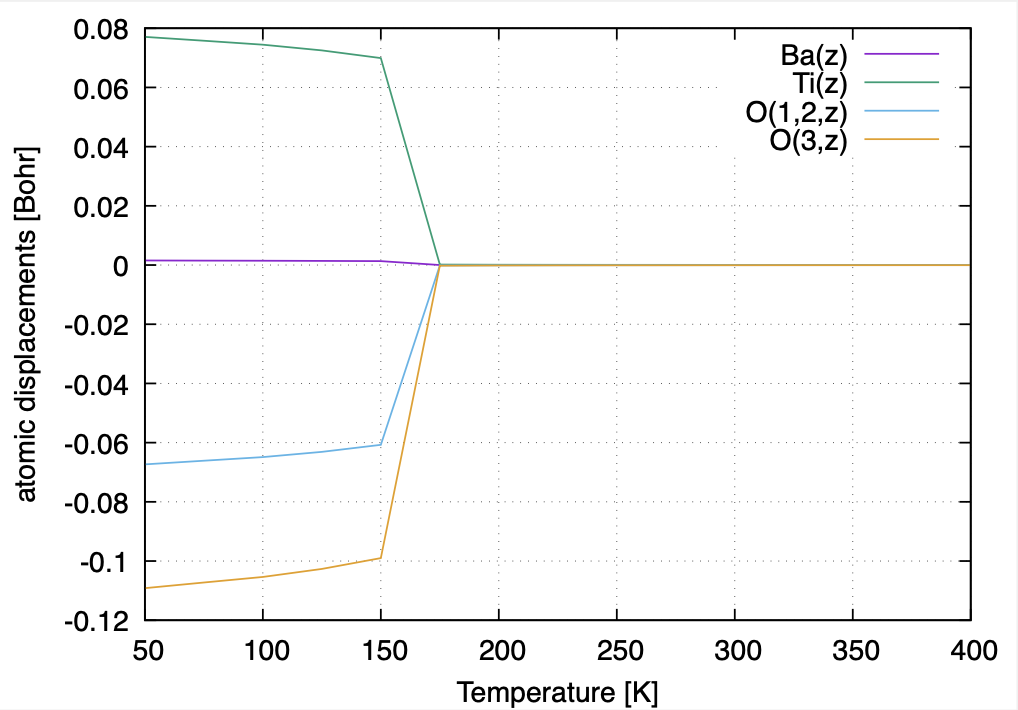
The \(T\)-dependence of the atomic displacements in cubic-tetragonal structural phase transition of BaTiO3.
The plot of the free energy can be obtained with
$ gnuplot plot_free_energy.plt
We can see that static energy \(U_0\) monotonically increases while the vibrational free energy \(F_{vib}\) decreases monotonically with temperature. Such changes in \(U_0\) and \(F_{vib}\) are especially drastic near the phase transition. The change of the total free energy is not as significant because the free energies of the two phases are equal at \(T_c\). Thus, we can see the competition between the enthalpy and the entropic terms in the \(T\)-dependence of the crystal structure.
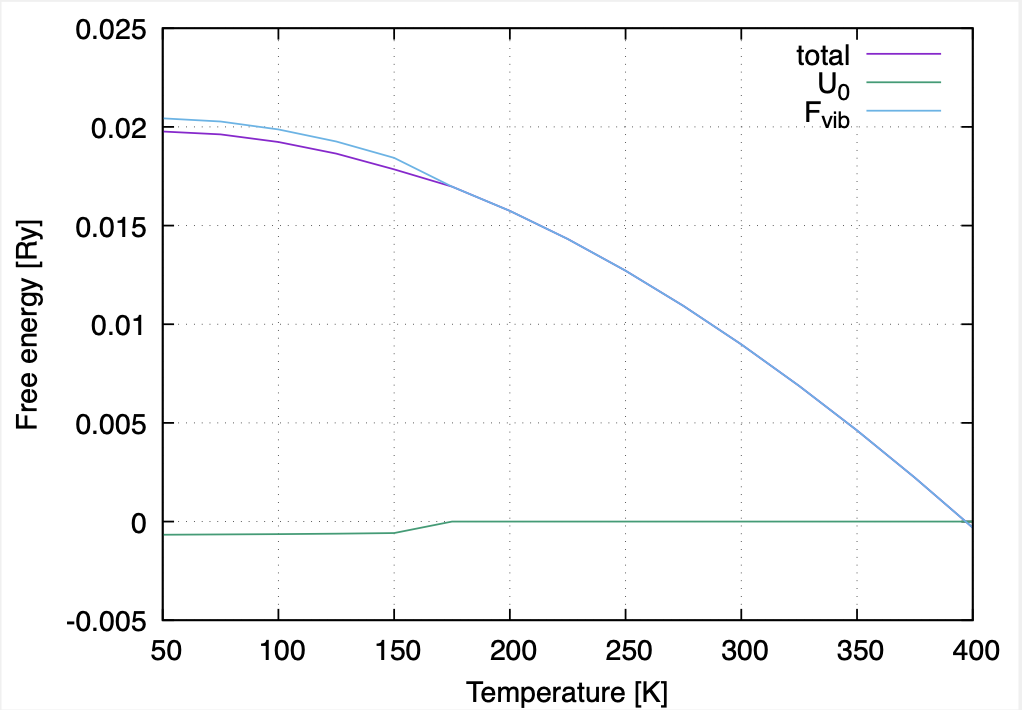
The \(T\)-dependence of the free energy in cubic-tetragonal structural phase transition of BaTiO3.
Note
We can estimate \(T_c\) more accurately from the crossing point of the free energies of different phases.
If we perform the cooling calculation without initial displacement, we will get the free energy of the cubic phase with lower temperatures.
If we perform the heating calculation, we may get the free energy of the tetragonal phase with higher temperatures.
Then, we can find the crossing point if the \(T\)-step (DT) is small enough.
If DT is too large to see the crossing point and the hysteresis,
we can extrapolate the free energy difference \(F_{cubic}-F_{tetra}\) from the low temperature to estimate \(T_c\).
Note
BaTiO3 shows a three-step structural phase transition between four different phases. For the other two phase transitions that occur at lower temperatures (tetragonal-orthorhombic and orthorhombic-rhombohedral transition), the symmetry of the low-\(T\) phases are not subgroups of the symmetry of the high-\(T\) phases.
In such cases, we recommend calculating cubic-orthorhombic and cubic-rhombohedral phase transitions separately and comparing the free energies because
The calculated hysteresis does not necessarily reflect the physics if the transition is strongly first-order.
The symmetry makes the calculation more stable and efficient. If we directly calculate the tetra-ortho transition, the symmetry used in the calculation is the common subgroup of the symmetry groups of these two phases, while we can take advantage of the full symmetry of the orthorhombic phase if we calculate the cubic-ortho transition instead.
Note
We will need to prepare additional inputs, the elastic constants, and the strain-harmonic-IFC coupling if we relax the unit cell as well. The strain-force coupling is not necessary for BaTiO3 because they are zero from symmetry.
Please see the Tutorial 7.8 for the details of the preparation of these inputs.